DFT Calculations on disaccharides:
New Insight into conformation and trans-O-glycosidic spin couplings
Roland Stenutza,
Francis Clorana,
Ian Carmichaelb,
and Anthony S. Serianni
a*
(a)Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN, USA 46556
(b)Radiation Laboratory, University of Notre Dame, Notre Dame, IN, USA 46556
Introduction
To understand the many biological roles of oligosaccharides, a knowledge of their
three-dimensional structure is required. One way to gain insight into the conformation
of an oligosaccharide is through the measurement of transglycosidic coupling constants.
Coupling constants have been shown to depend on the geometry of the molecule, mainly the
torsion angle between the coupled nuclei but also bond lengths and bond angles and
substitution pattern.
Using ab initio quantum mechanical methods, coupling constants can be calculated.
Herein we apply Density Functional Theory (DFT), a quantum mechanical analysis based on
electron density, to disaccharide models to optimize their geometries by minimizing the
potential energy of the system. From the optimized geometries, J coupling values
can be determined by calculating the Fermi-contact component of the indirect spin-spin
coupling constant. The accuracy of these Jvalues can be assessed through comparison
of the computed J values with experimental data. As the computed
JCH values were somewhat smaller and the JCC
slightly larger than the experimental values, adjustments of +5% and -10% respectively
were applied.
In this study, the geometries of some models of β-(1→4) linked disaccharides were
optimized as a function of the glycosidic torsion angle, ΨH.
The geometries were then used to calculate two-bond and three-bond
trans-O-glycosidic J couplings to determine the usefulness of
Density Functional Theory to the analysis of larger molecular systems, such as
disaccharides.
Experimental
All ab initio calculations were performed with a modified version of the GAUSSIAN 94
program using the UB3LYP/6-31G* basis set [6] and Density Functional
Theory (DFT) to account for electron correlation effects. The Fermi-contact contribution
to the scalar coupling constants were calculated using the finite pertubation method.
The basic disaccharide mimics used in these calculations are shown in Scheme 1.
Scheme 1
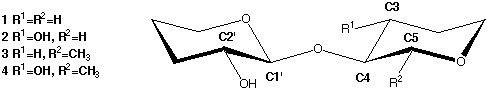
|
The starting geometries of the ΦH-torsions (H1'-C1'-O4-C4) were set to
+60° to give the rotamers stabilized by the exoanomeric effect. The
H2'-C2'-O2'-HO2' torsion was arbitraily set to +60° and, when applicable, the
C2-C3-O3-HO3 torsion set to 180°. All three staggered rotamers around the
ΨH torsion (C1'-O4-C4-H4) were optimized but only in the case of
compound 1 did this result in two different gauche conformers. In compounds
2, 3 and 4 both gauche (~60°) starting geometries gave the
same optimized structure. This is in agreement with results from molecular
mechanics/molecular dynamics and NMR which suggest only one "syn"
(ΨH~0°) geometry with significant flexibility.
On completion of the geometric optimizations, J calculations were performed for the
pathways indicated below in Scheme 2.
Scheme 2
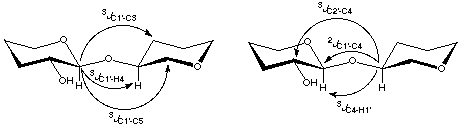
|
Results and Discussion
Geometry
From the three staggered starting geometries only two final geometries are obtained for
compounds 2-4 after geometry optimization: anti
(ΨH~180°) and syn (ΨH~-20°). The
values for ΨH shown in table 1 depart considerably from the idealized
staggered torsion angles of ~60°.
Table 1
| Compound |
syn (ΨH~0°) |
anti (ΨH~180°) |
|
ΦH | ΨH |
ΦH | ΨH |
| 1a | +47° | -17° | +32° | +171° |
| +43° | +22° | | |
| 2 | +32° | -25° | +18° | +180° |
| 3 | +44° | -19° | +52° | -159° |
| 4 | +32° | -22° | +17° | -179° |
| β(1→4)b | +23° - +60° | -51° - +12° |
| |
a) two syn-structures
b) range for 35 X-ray structures of β(1→4) linked disaccharides in the Cambridge crystallographic database
|
It is interesting to note that, in the unsubstituted compound (1), the two gauche rotamers must be separated by an energy barrier since they give different optimized geometries. In all cases, the syn geometry was lower in energy than the anti geometry by 2.4-3.2 kcal/mol. Thus the anti conformation is not necessarily inaccessible but much less favored than the syn conformation.
Trans-O-Glycosidic 2JCOC Values
In the optimized structures the glycosidic 2JCOC were calculated.
According to the empirical projection resultant method [4],
the size of 2JCOC depends only on ΦH and should be ~-2 Hz
for all the compounds. This is consistent with the results obtained from the Fermi-contact calculations which gave values of -1.9 to -2.5 Hz (average -2.0 Hz). These results encourage the use of DFT for future two-bond glycosidic J coupling analyses.
Trans-O-Glycosidic 3JCOCH Values
There are two glycosidic 3JCOCH of interest. Table 2 lists the computed coupling constants as well as their associated dihedral angles.
Table 2
Calculated Three Bond CH Couplings in Model Disaccharides
| Structure | 3JC1'-H4 |
3JH1'-C4 |
| 1 | syn (sc-) | 6.1 (-17°) | 4.3 (48°) |
| syn (sc+) | 5.9 (22°) | 4.2 (43°) |
| anti | 8.3 (171°) | 4.5 (32°) |
| 2 | syn | 5.5 (-26°) | 5.4 (32°) |
| anti | 9.0 (180°) | 4.6 (18°) |
| 3 | syn | 6.1 (-19°) | 4.5 (44°) |
| anti | 8.4 (-160°) | 2.4 (52°) |
| 4 | syn | 5.7 (-22°) | 5.1 (32°) |
| anti | 9.0 (-179°) | 4.4 (16°) |
|
Although not quantitative, these computed J couplings are of the same size as those reported
experimentally. It has also been noted that 3JCOCH constants display a
Karplus-like dependency on the torsion angle between the coupled atoms. Figure 1
is a plot of the computed J couplings versus their associated dihedral angles. Included
in this plot for comparison is an experimentally derived Karplus curve for
3JCOCH reported previously [7].
It is difficult to ascertain whether or not the DFT results support the empirical Karplus relationship of 3JCOCH because of the absence of data for the dihedral angles around ~90°. Efforts are being made to measure theoretical J couplings over the entire torsional range for this three-bond pathway.
Figure 1
Calculated 3JCOCH
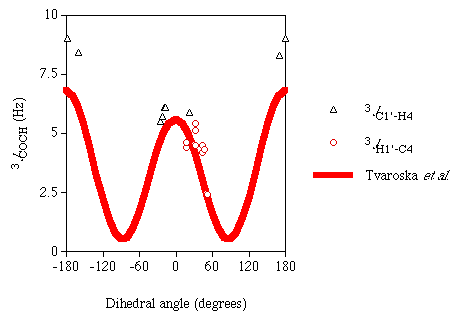
|
Trans-O-Glycosidic 3JCOCC Values
The other 3J of interest across the glycosidic linkage are the three carbon-carbon couplings. Table 3 lists the computed coupling constants as well as their associated dihedral angles between the coupled nuclei.
Table 3
Calculated Three-Bond CC Couplings in Model Disaccharides
| Compound | 3JC1'-C3 |
3JC1'-C5 |
3JC2'-C4 |
| 1 | syn (sc-) |
0.0 (105°) | 3.3 (-135°) | 3.8 (168°) |
| syn (sc+) |
2.2 (141°) | 0.0 (-99°) | 3.4 (164°) |
| anti |
0.0 (-72°) | 1.7 (55°) | 3.3 (152°) |
| 2 | syn |
0.0 (95°) | 4.4 (-144°) | 3.4 (153°) |
| anti |
1.0 (-65°) | 1.4 (63°) | 2.3 (137°) |
| 3 | syn |
0.0 (102°) | 3.6 (-137°) | 3.7 (164°) |
| anti |
3.2 (-42°) | 0.0 (86°) | 3.7 (172°) |
| 4 | syn |
0.0 (98°) | 3.9 (-140°) | 3.3 (153°) |
| anti |
1.2 (-64°) | 1.3 (65°) | 2.3 (137°) |
|
The values for these computed J couplings of similar magnitude as those recently reported experimentally in this laboratory, although not quantitative. As with the 3JCOCH values, magnitudes for 3JCOCC are reported to express a Karplus dependency on the dihedral angle between the two coupled nuclei. Figure 2 is a plot of the computed carbon-carbon couplings versus the associated dihedral angles. Included in this plot is a graph of the idealized empirical Karplus curve for this coupling pathway recently determined in this laboratory [8].
Figure 2
Calculated 3JCOCC
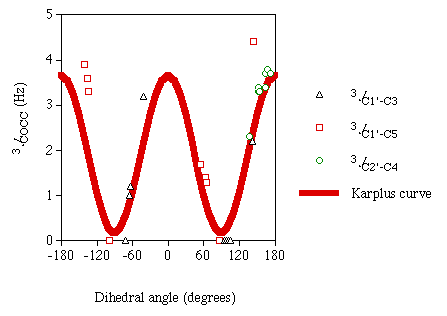
|
In this plot one can see a Karplus-like dependency of the
3JCOCC values on the associated dihedral angles. In the
dihedral angle range of 140° to 170°, a scattering of the data points from an
idealized curve is observed. This scattering may be explained by substituent effects (O5
in plane) along the J coupling pathway. If there were substituent effects on the
3JCOCC pathway, which is seen in
3JHH pathways, one might expect to see these effects
amplified on the steeper part of the Karplus curve. Despite these deviations, the computed
3JCOCC show good agreement with the Karplus equation. These
results are encouraging and justify future application of DFT to analyze other model
disaccharides.
Summary
-
In the DFT analysis of disaccharide models 2-4 where ΨH
was initially varied, two optimized geometries were obtained from the three staggered
starting geometries: one geometry where ΨH was ~180° (anti) and
the other geometry where ΨH was ~-20° (syn). It was determined in
each case that the anti geometry was a higher energy structure than the syn geometry by
2.4-3.2 kcal/mol.
-
2JCOC couplings across the glycosidic linkage were calculated for all optimized structures. Couplings of approximately -2 Hz were calculated, which is in good agreement with the results of the projection resultant method.
-
3JCOCH across the glycosidic linkage were calculated for all
optimized structures. The 3JCOCH were similar to those
measured experimentally [7]. Due to the absence of models with dihedral
angles around ~90°, the 3JCOCH couplings could not be
used to test the presence of a Karplus relationship in these model structures.
- 3JCOCC couplings were calculated for all optimized structures. The 3JCOCC were close to those measured experimentally on similar compounds. Despite some scatter for dihedral angles around 180°, the relationship of the 3JCOCC to the associated dihedral angles was in good agreement with an experimental Karplus relationship reported recently for the 3JCOCC pathway [8].
References
-
Levine, I.N.; Quantum Chemistry; Prentice-Hall, Inc.: Englewood Cliffs, NJ, 1991.
-
Lebanowski, J.K.; Andzelm, J.W. (eds.);Density Functional Methods in Chemistry; Springer-Verlag: New York, NY, 1991.
-
Haasnoot, C.A.G.; de Leeuw, F.A.A.M.; Altona, C.; Tetrahedron 36, 2783-2792 (1980).
-
Church, T.; Carmichael, I.; Serianni, A.S.; Carbohydr. Res. 280, 177-186 (1996).
-
Carmichael, I. J. Phys. Chem.; 97, 1789-1792 (1993).
-
Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Gill, P.M.W.; Johnson, B.G.; Robb, M.A.; Cheeseman, J.R.; Keith, T.; Petersson, G.A.; Montgomery, J.A.; Raghavachari, K.; Al-Laham, M.A.; Zakrzewski, V.G.; Ortiz, J.V.; Foresman, J.B.; Peng, C.Y.; Ayala, P.Y.; Chen, W.; Wong, M.W.; Andres, J.L.; Replogle, E.S.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Binkley, J.S.; Defrees, D.J.; Baker, J.; Stewart, J.P.; Head-Gordon, M.; Gonzalez, C.; Pople, J.A.;Gaussian 94, Revision B.3; Gaussian, Inc.: Pittsburgh, PA, 1995.
-
Tvaroska, I.; Hricovíni, M.; Petráková, E.; Carbohydr. Res. 189, 359-362 (1989).
-
Bose, B.; Zhao, S.; Stenutz, R.; Cloran, F.; Bondo, P.; Bondo, G.; Hertz, B.; Carmichael, I.; Serianni, A.S.; J. Am. Chem. Soc., submitted for publication.
|
|