4.1 Introduction
Butyrivibrio fibrisolvens is an anaerobic bacterial species commonly found in the intestine of herbivorous mammals. The species can be divided into two distinct taxonomic groups depending on the presence or absence of an unusual acidic sugar, 6-O-[(R)-1-carboxyethyl]-D-galactose (1),79 in their extracellular polysaccharide. Another acidic sugar, 4-O-[(S)-1-carboxyethyl]-L-rhamnose,80,81 was present in only one of the strains examined. Sugars substituted with carboxyethyl groups are also produced by some other bacteria.82| methyl 6-O-[(R) -1-carboxyethyl] -α-D-galactopyranside (1) | methyl 4-O-[(S) -1-carboxyethyl] -α-L-rhamnopyranoside |
![methyl 6-O-[(R)-1-carboxyethyl]-a-D-galactopyranside](gif/galact.gif) |
![methyl 4-O-[(S)-1-carboxyethyl]-a-L-rhamnopyranoside](gif/rhamn.gif) |
The relative configuration of the carboxyethyl group can then be determined by NOE measurements. As methyl glycosides are preferable for the spectroscopic studies and allow a direct comparison with native polysaccharides we chose to prepare them rather than the free sugars 85 or carboxyl reduced derivatives.86
4.2 Preparation of carboxyethyl sugars
Alkylation of sugar derivatives with optically pure α-chloropropionic acid had been reported previously in connection with the synthesis of 2-amino-3-O-[(R)-1-carboxyethyl]-2-deoxy-D-glucose (muramic acid) 87 and 3-O-(1-carboxyethyl)-L-rhamnose.85 By this approach the separation of diastereomers was avoided.The (R)- and (S)-α-chloropropionic acids 88 were prepared by diazotation of (R)- and (S)-alanine respectively in 3M HCl. Initially the highly strained α-lactone of lactic acid is formed which is opened by chloride ion to give chloropropionic acid with overall retention of configuration.
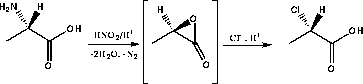
|
In order to make an unambiguous assignment of the H-6 protons stereospecifically deuterated derivatives were prepared (scheme 4.2). Photochemical bromination of 2,3,4-tri-O-benzoyl-1,6-anhydrogalactose resulted in the exchange of the exo-hydrogen on C-6, i.e. H-6proS, for bromine. After purification the bromide was subjected to radical mediated dehalogenation using tributyltin deuteride whereby deuterium was introduced with the same stereochemistry.89 After Zemplén deacetylation and benzylation the 1,6-anhydroring was opened by acetolysis giving a mixture of diacetates. Treatment of the diacetates with methanolic hydrogen chloride resulted in simultaneous deacetylation and Fischer glycosylation. After chromatographic separation the pure α-anomer could be obtained. The α-glycoside was then alkylated and deprotected as described for the undeuterated compounds.
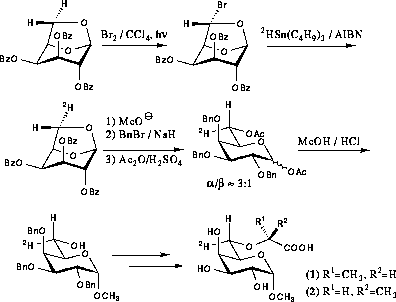
|
4.3 NMR studies
The assignment of the 1H-NMR signals of the rhamnose derivatives was straightforward using 1H,1H-COSY spectra and subsequently the 13C-signals using 13C,1H correlated spectra. The assignment of the spectra of compounds 1 and 2 was complicated by the close proximity of several 1H- as well as 13C-NMR resonances. The signal for C-6 could be assigned using DEPT experiments and this in turn allowed the assignment of the H-6 signals. In an attempt to resolve the remaining uncertainties a DIS 90 experiment was performed.Normally spectra are recorded in D2O which causes a slight but noticeable isotope effect on the hydroxyl-bearing carbons and their neighbours. This isotope effect can be measured by comparing spectra recorded in D2O and in 80-90% H2O/D2O . By comparing the effects on methyl α-D-galactopyranoside with those on 1 and 2 the assignments for C-4 and C-5 of compound 2 could be determined. The diastereomeric hydroxymethyl protons could be assigned since the H-6proS resonances are absent in the deuterated compounds.
All four compounds showed typical substitution shifts in 13C-NMR (α-C downfield 7-10 ppm, β-C upfield 0.5-2.5 ppm). The effects on the 1H-NMR spectra were minor. The differences between the pairs of diastereomers were small and an assignment of configuration in a polysaccharide based on NMR only would be difficult.
4.4 Conformational analysis
The C5-C6 bond of hexopyranoses is known to be very flexible and by the substition at O6 two additional flexible bonds are introduced. Since the same degrees of freedom are present in 6-linked saccharides, such as dextran, and hence the same computational and experimental difficulties should be encountered, it was of interest to attempt to determine the conformations of 1 and 2.The three exocyclic torsions are labelled φ' (H2'-C2'-O6-C6), ψ' (C2'-O6-C6-C5) and ω (O5-C5-C6-O6), respectively, in analogy with 6-linked disaccharides.
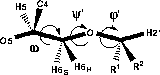
| |||||
| Pg+ | Pt | Pg- | ||
| 1 | φ' | -a (30) | 40 (2) | -a (68) |
| ψ' | 60 (14) | 0 (68) | 40 (18) | |
| ω' | 57 (68) | 26 (25) | 18 (7) | |
| 2 | φ' | -a (92) | 32 (0) | -a (8) |
| ψ' | 55 (2) | 0 (60) | 45 (38) | |
| ω' | 62 (95) | 26 (3) | 12 (2) | |
| Me α-D-Galp | ω | 47 | 39 | 14 |
| a) Experimental data does not allow for the separate calculation of populations in the gauche states. | ||||
Across the φ'-torsion there is only one heteronuclear coupling that can be measured, that of H2' to C6. The values obtained both for 1 and 2 suggest that this torsion is about one third trans, as would be expected in the absence of any strong conformational preference (table 4.1). Although the rotamer population distributions should be similar to those calculated from the coupling constants, it must be kept in mind that the accuracy of Karplus equation for 3JCH is significantly lower than that for 3JHH as the former was developed for glycosidic linkages and does not take electronegativity effects into account. 91 Coupling constants alone do not provide any information as to the mutual dependence of the individual torsions. Such information could be available from NOE measurements but this requires well resolved resonances. Although calculated populations for 1 and 2 are very similar the measured differences in coupling constants are significant.
| JH5,H6R | JH5,H6S | JH6S,C2' | JH6R,C2' | JC6,H2' | |
| 1 | 7.1 | 4.6 | 3.7 | 4.9 | 3.7 |
| (7.7) | (3.8) | (2.7) | (2.9) | (3.1) | |
| 2 | 7.6 | 4.6 | 4.2 | 4.8 | 3.3 |
| (9.7) | (1.5) | (2.8) | (3.2) | (2.6) | |
| Me α-D-Galp70 | 7.8 | 6.0 |
| 3JHH= P1cos2θ+ P2cos θ+ P3+ ΣΔχi{ P4+P5cos2 (ζθ+P4|Δχi|)} [1]92 |
| where P1-P6 are constants, Δχi the relative electronegativities (rel. to H) of the substituents, ζ a constant depending on the orientation of a substituent and θ the H-H-torsion angle. |
| 3JCOCH= 5.7cos2θ-0.6cos θ+0.5 [2]67 |