2.1 Introduction
The structural analysis of oligo- and polysaccharides poses some unique
problems since, unlike the amino acids in peptides or nucleosides in DNA,
sugar residues can have different ring structures, be linked at several
different positions and may form not only linear, but also branched and
cyclic structures. Most bacterial polysaccharides are regular, i.e.
composed of repeating units, and may contain branches. There are other
carbohydrate polymers which are dendritic or irregular but they will not
be considered here as their structure can not be uniquely defined but
requires statistical treatments.
The structural analysis of oligo- and polysaccharides by chemical
methods 8
alone may be tedious and often requires large amounts of
material. Modern analytical methods such as NMR-spectroscopy and mass
spectrometry 9-11
have made it possible to reduce both the amount of
substance and the time required to perform analyses. The use of these new
sensitive methods has also made it possible to identify components which
are easily lost or transformed during chemical analysis. In particular the
ability of NMR-spectroscopy to determine the anomeric configuration has
led to revision of older structure proposals. Despite this, chemical
analysis remains the preferred method for component and linkage
analysis.
2.2 NMR-spectroscopy 12,13
There are several advantages in using NMR-spectroscopy in structure
analysis. Labile components, such as diaminosugars or O-acetyl groups,
which may be destroyed or transformed during chemical manipulations
can be detected. Ring size and anomeric configuration may be difficult to
determine with other methods. In order to use NMR-spectroscopy for
sequence determination it is, however, necessary to assign individual
resonances which in itself may require several time-consuming
NMR-experiments.
Several computer programs have been developed to aid in structure
determination using NMR-spectroscopy and to speed up sequence
determination. There are two different strategies:
2.2.1 Database search
If a compound is already known and its spectrum has been recorded it
may be identified by direct comparison with a reference spectrum. No
chemical manipulations are required and hence only a minimum of
substance is needed. This approach has been successfully used in the
structure determination of glycoprotein 14
and xyloglycan
15
derived oligosaccharides based on 1H-NMR data.
The use of databases has one
disadvantage - it requires a reference spectrum. The identification of new
compounds is therefore difficult, although substructures may be
recognised.
2.2.2 Spectrum simulation
If NMR spectra are calculated from the chemical shifts of the constituent
sugars using some rules, the spectra of new structures can be
approximated. 16-19
The glycosylation of a monosaccharide causes changes
in its NMR spectrum referred to as glycosylation shifts. Their size
depends on the geometry of the glycosidic bond. Using glycosylation
shifts for different linkages and the chemical shifts of the
monosaccharides, the calculation of a spectrum for any sequence and
substitution pattern is possible. Providing that only short distance
interactions are present additivity of glycosylation shifts is obeyed.
Notable exceptions are residues where vicinal substitution, e.g. branch
points, causes steric interactions not present in the model disaccharides.
This can be compensated to some extent by including corrections based on
trisaccharide fragments.
Disadvantages of this approach are that a large set of assigned di- and
trisaccharide fragments is required to obtain glycosylation shifts and that
the simulated spectra are less accurate than spectra in a database.
Scheme 2.1: Calculation of chemical shifts
| 1) Chemical shifts are taken from the monosaccharide |
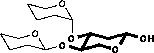 |
| 2) Glycosylation shifts are added |
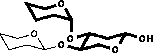 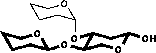 |
| 3) In the case of vicinal substitution corrections are added |
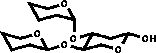 |
Using results from component and linkage analysis all possible
permutations of sequences and anomeric configurations are generated.
The calculated spectra of all generated structures are compared with
experimental data and ranked according to fit. If the spectrum simulation
performs well there will be at least one calculated spectrum which shows
good agreement with experiment allowing other structures to be rejected.
A comparison between the simulated and experimental spectra gives not
only the structure but also the assignments for all signals, something
which is advantageous when several plausible structures are to be
discriminated by additional NMR experiments.
2.3 Enhancement of the CASPER program (Paper I)
CASPER is a computer program which automates the generation of trial
structures and the calculation of chemical shifts according to the above
scheme. It has previously been successfully applied to linear 17
and branched 18
oligo- and polysaccharides. Structure determinations have
required complete sets of experimental 13C-NMR chemical shifts, in
addition to information on the component sugars and their linkages. The
inclusion of coupling constants of the anomeric protons can be used to
reduce the number of simulated structures. Some of the limitations of
earlier versions have now been addressed:
- The format of the database containing the chemical shifts
of the monosaccharide residues, glycosylation shifts and
branch point corrections has been changed to facilitate
database maintenance and extension.
- The possibility to simulate multiply branched structures
has been added.
- The use of incomplete sets of chemical shifts is now
possible so that poorly resolved spectra may be used.
The enhanced program was tested on one oligo- and three
polysaccharides of known structure.
2.3.1 Simulation using a reduced number of resonances
The complete 13C-NMR spectrum of the O-polysaccharide of the LPS
from Shigella flexneri type 4a 19
was used as a starting point since the
spectrum is well simulated by CASPER. Omission of resonances from the
experimental spectrum were made at random before it was used as input
for structure determination. Successive removals of signals diminishes the
error of the fit for all the simulated structures making them increasingly
difficult to discriminate. Although no rigorous treatment was attempted,
the results suggest that a substantial number of signals may be omitted
before it becomes uncertain which structure is correct.
2.3.2 Multiply branched polysaccharides
Two doubly branched polysaccharides were investigated; the
O-polysaccharide of the LPS from an Aeromonas caviae strain
20
and the
capsular polysaccharide from Klebsiella K8,52,59.
21
In the A. caviae
polysaccharide both branches are attached to the same residue in the
backbone, in the Klebsiella CPS to different residues. Of the 30 signals
expected in the spectrum of the Klebsiella CPS, only 28 were easily
identified and therefore a reduced set of experimental chemical shifts was
used. In both the above cases the correct structure was ranked highest but
the fit was not as good as for the S. flexneri polysaccharide. The largest
errors are confined to the residues around the branch points where
deviations from additivity due to steric crowding are to be expected.
Extending the database with more corrections for branching will help to
reduce this problem.
2.3.3 An oligosaccharide of the high-mannose type 22
Oligosaccharides from glycoproteins are generally highly branched and
composed of only a few different sugars with similar substitution
patterns. This could make it difficult to distinguish between the different
structures. As the simulated spectum showed a good fit it was possible to
identify the correct structure for an octasaccharide containing five
α-mannose residues. Since the sensitivity of
13C-NMR spectroscopy is low
and the spectra of the structures are similar a database approach using
1H-NMR
is to be preferred for this type of structures. It does, however,
demonstrate that extensive branching, per se, is not an obstacle.
2.4 The structure of the Klebsiella K52 CPS (Paper II)
A partial structure of the capsular polysaccharide of Klebsiella K52 had
previously been determined by methylation analysis and partial acid
hydrolysis 23
but the anomeric configurations of the residues remained unknown.
Fig 2.1 Structure of Klebsiella K52 CPS
→4)-αLRha-(1→3)-βDGal-(1→2)-αLRha-(1→4)-βDGlcA-(1→3)-αDGal(1→
2
↑
1
αDGal
|
Using the 1JCH
values of the anomeric carbons it would be easy to
distinguish between those that have an equatorial (α,
1JCH≈170 Hz) and those with an axial (β,
1JCH≈160 Hz) proton.24
It is also possible to use 3JHH
between H1 and H2 which is larger in a β-gluco-linkage (≈8 Hz) than
in an α-gluco-linkage (≈4 Hz). 25
The difference between α and β is much
smaller in the manno case (both < 2 Hz). To assign α or β configuration
to the different residues it is necessary to have the assignment of the NMR
resonances. The assignments are also required for sequence determination
using inter-residue NOEs or long-range heteronuclear couplings
(3JCOCH).
Assignments can be obtained by traditional NMR methods, i.e. from
1H,1H-COSY and
13C,1H-correlated spectroscopy.
If some resonances are
absent, as in this case, this is not easily accomplished.
Three signals were not readily discernible in the
13C-NMR spectrum of
the CPS probably because of low intensity or overlap of resonances. The
remaining signals were used as input to CASPER together with the results
of component and linkage analysis. The previously suggested sequence
had the best fit, although the difference between the first two structures
was too small to make an unambiguous selection of the correct structure.
The highest ranked structure suggestions differed in sequence rather than
in anomeric configuration. In order to verify the suggested structure
(fig 2.1) HMBC and NOE experiments were performed, giving an
independent confirmation of the connectivity between the residues. It was
concluded that the original structure was correct and only had to be
amended with the anomeric configurations.
2.5 Conclusions
The use of computer programs such as CASPER can speed up structure
determination significantly. After the latest additions to the program it
should be possible to simulate the spectrum of any oligo- or
polysaccharide structure provided that the necessary glycosylation shifts
are known. In cases where the experimental data are incomplete it is still
possible to obtain meaningful results. The better dispersion of resonances
in 13C-NMR spectra makes it more suitable for spectrum
simulations than 1H-NMR spectra where most resonances overlap even at
very high field. The higher sensitivity of 1H-NMR spectroscopy makes it
ideal for the reporter group approach,26
i.e. the use of structure specific signals, or
database approaches. A weakness of CASPER is the database. The number
of di- and trisaccharides investigated is not sufficient to cover all the
structures of biological interest. The NMR spectra of many compounds,
both oligo- and polysaccharides, have been published and a logical next
step in the development of the program would be to allow for the
inclusion of these and thereby increase both the accuracy and scope of the
program.
|
|